INCLUDE HTP DESeq2 Tutorial: Step-by-step instructions
This tutorial project uses high-throughput (HTP) research data from the INCLUDE Data Hub, configured for use on the CAVATICA platform. The analysis covers high-throughput differential expression analysis using DESeq2 and Gene Set Enrichment Analysis (GSEA) in R. The analysis specifically focuses on testing for differential gene expression in high-throughput PAXgene Whole Blood RNA samples generated from paired-end PolyA+ globin-depleted libraries.
This project builds computational and data science capacity within the INCLUDE Project and the broader Down Syndrome research community. It was inspired by materials developed by Matthew Galbraith for the Data Science for Developing Scholars in Down Syndrome Research (DS3) program; see the GitHub repository and the associated publication for more details.
To learn more about this project, or how to adapt it to your own research, please reach out to [email protected].
What the INCLUDE DESeq2 Tutorial Covers
This tutorial guides you through the statistical modeling of RNA-seq count data using the negative binomial distribution via DESeq2 to identify differentially expressed genes.
It also covers GSEA Hallmark analysis to evaluate whether curated hallmark gene sets - representing well-defined biological states or processes - are significantly enriched in a ranked list of genes, revealing coordinated pathway-level changes between conditions.
Set up the project and run the R script in Data Studio
To perform the INCLUDE HTP DESeq analysis, you’ll need to make your own copy of the Public Project and then execute the R script in a Data Studio session.
Public Projects contain all necessary files, instructions, and tools necessary to perform the specified analysis(es). However, the Public Project serves as a repository for all users, and as such, is not editable.
1. Copy the public project
1.1. Click Public Projects on the top navigation bar.
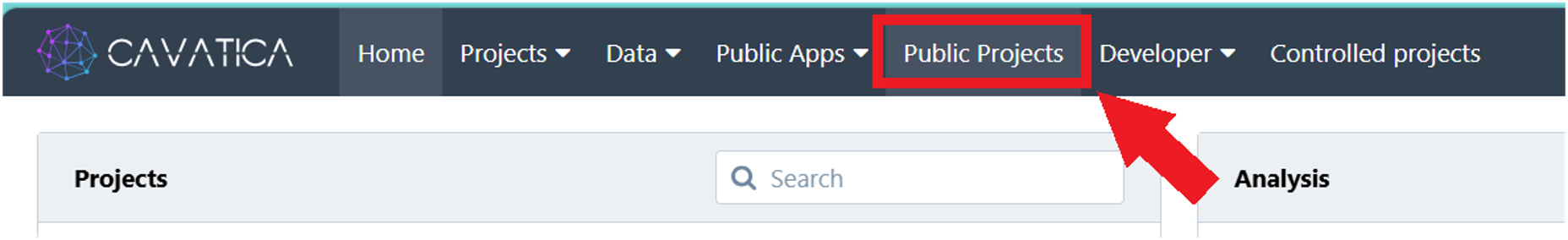
1.2. Click the "INCLUDE HTP DESeq2 Analysis".
1.3. Click the ‘i’ icon next to the public project title, and then click Copy project. The ‘Copy Project’ window will appear.
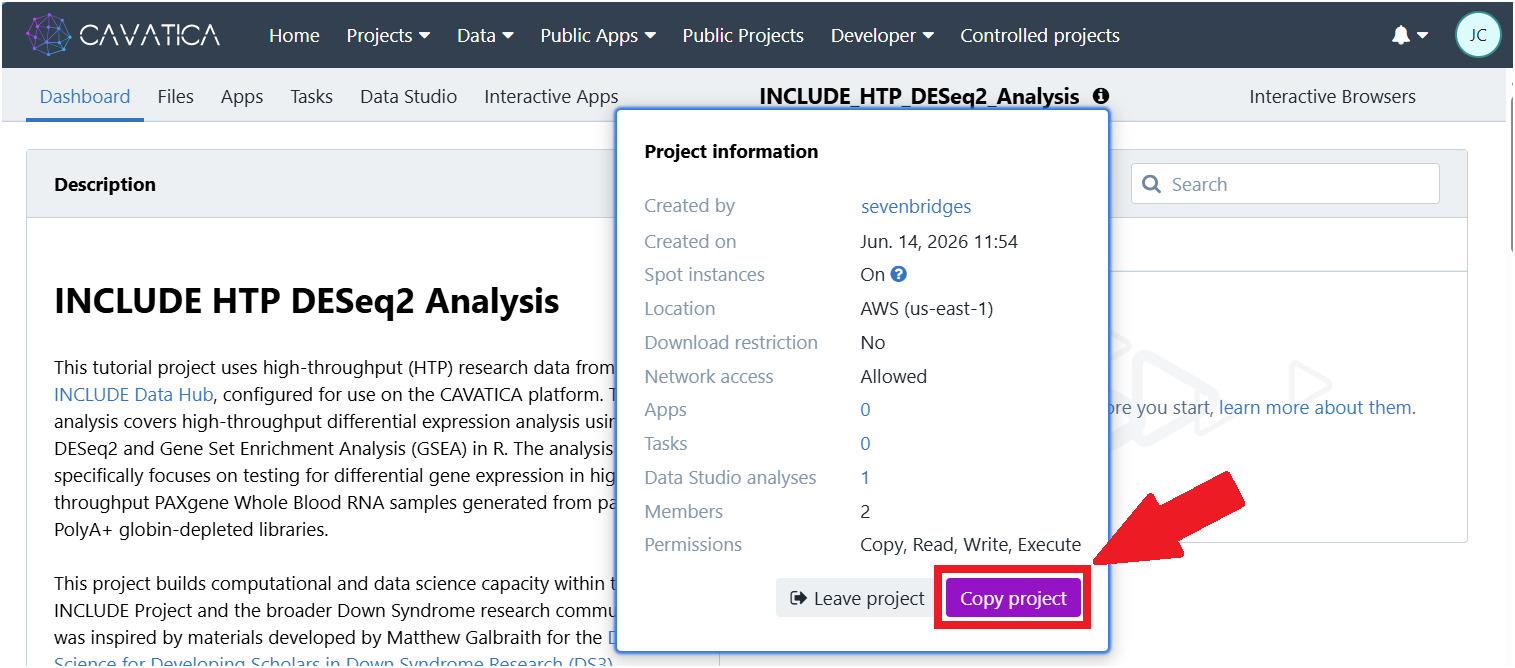
1.4. Give your project a unique name. 1.5. Under "Billing group", select your pilot funds group. 1.6. Click Copy (bottom right).
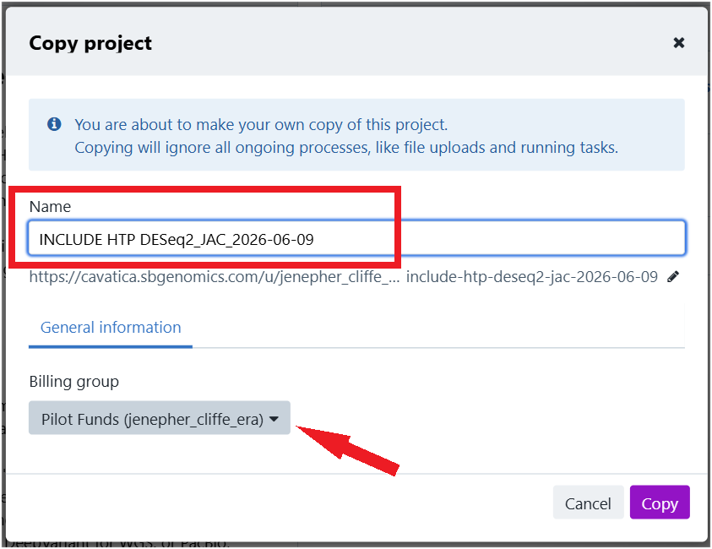
2. Set up environment and start RStudio
2.1. Copy the project by clicking the icon next to the project name, and selecting "Copy project".
2.2. Open Data Studio session: Navigate to Data Studio along the top gray banner of your copy of the project and click the purple start icon beside INCLUDE HTP DESeq2 Analysis.
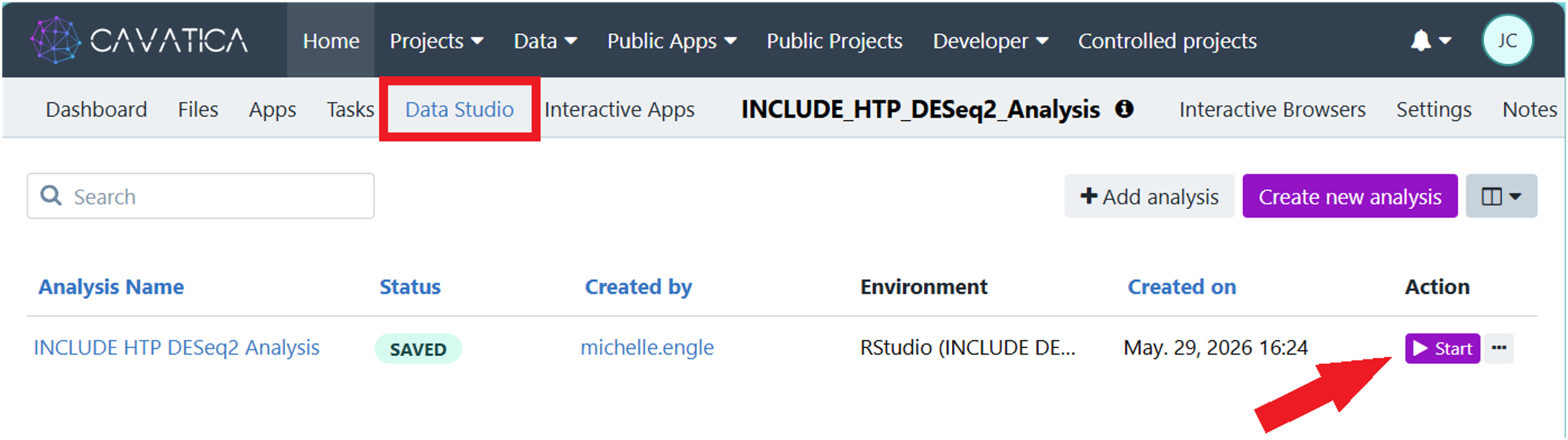
Note that you will have to wait a few minutes while SB initializes the Data Studio virtual machine.
2.3. Open the R script in RStudio by clicking on HTP_RNASeq_DESeq2_v3.2.R in the Files window (lower right pane).
3. Explore the main R Script
3.1. Review and run the analysis step-by-step by clicking the Run icon above the console window.
Comments in the code explain each step as you run it.
4. Clean up and close Data Studio
4.1. Clean up cache before stopping the analysis
This ensures a faster save process when stopping your analysis session
Run the following command in the terminal
sudo rm -rf ~/.cache/R/ && rm -rf ~/.local/share/4.2. Stop the analysis once everything is complete.
This will take a few minutes, as SB syncs the files in the /output-files directory to the project Files.
💡Saving outputs to project files
Output files generated during your analysis are stored in the Data Studio environment and are not accessible outside your private DS session. Note that the last part of the R script copies the files from/sbgenomics/workspace/to the/sbgenomics/output-filedirectory, which is automatically synced to the project when you stop the Data Studio analysis.Once the analysis is stopped, these files will be available in the Files tab of the project (along the gray navigation bar).
Customization (advanced resources)
Note that we have included advanced options for customizing the data Studio environment in the “advanced_customization folder inside the analysis directory.
Updated about 1 month ago